Spike protein visualisations
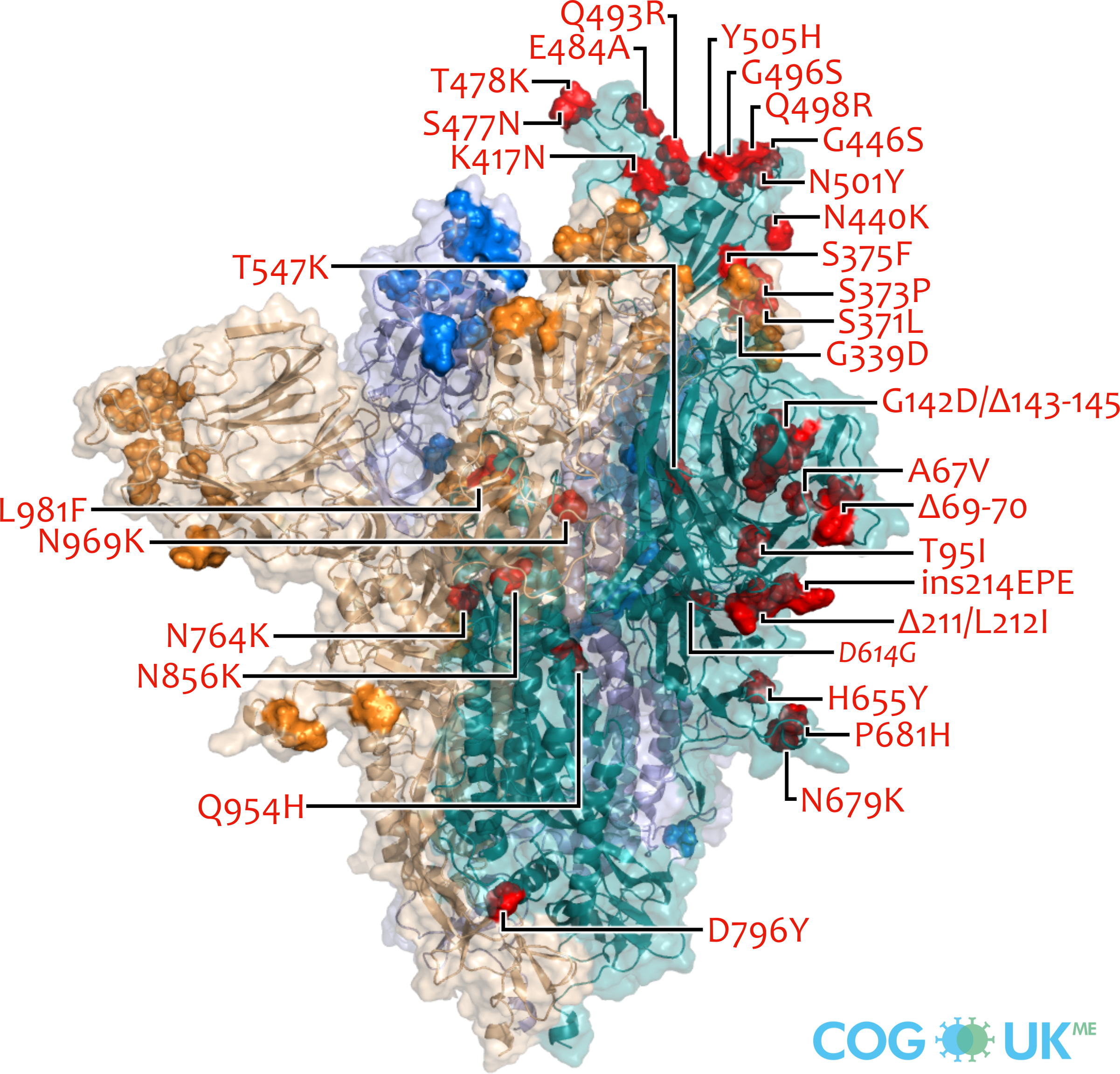
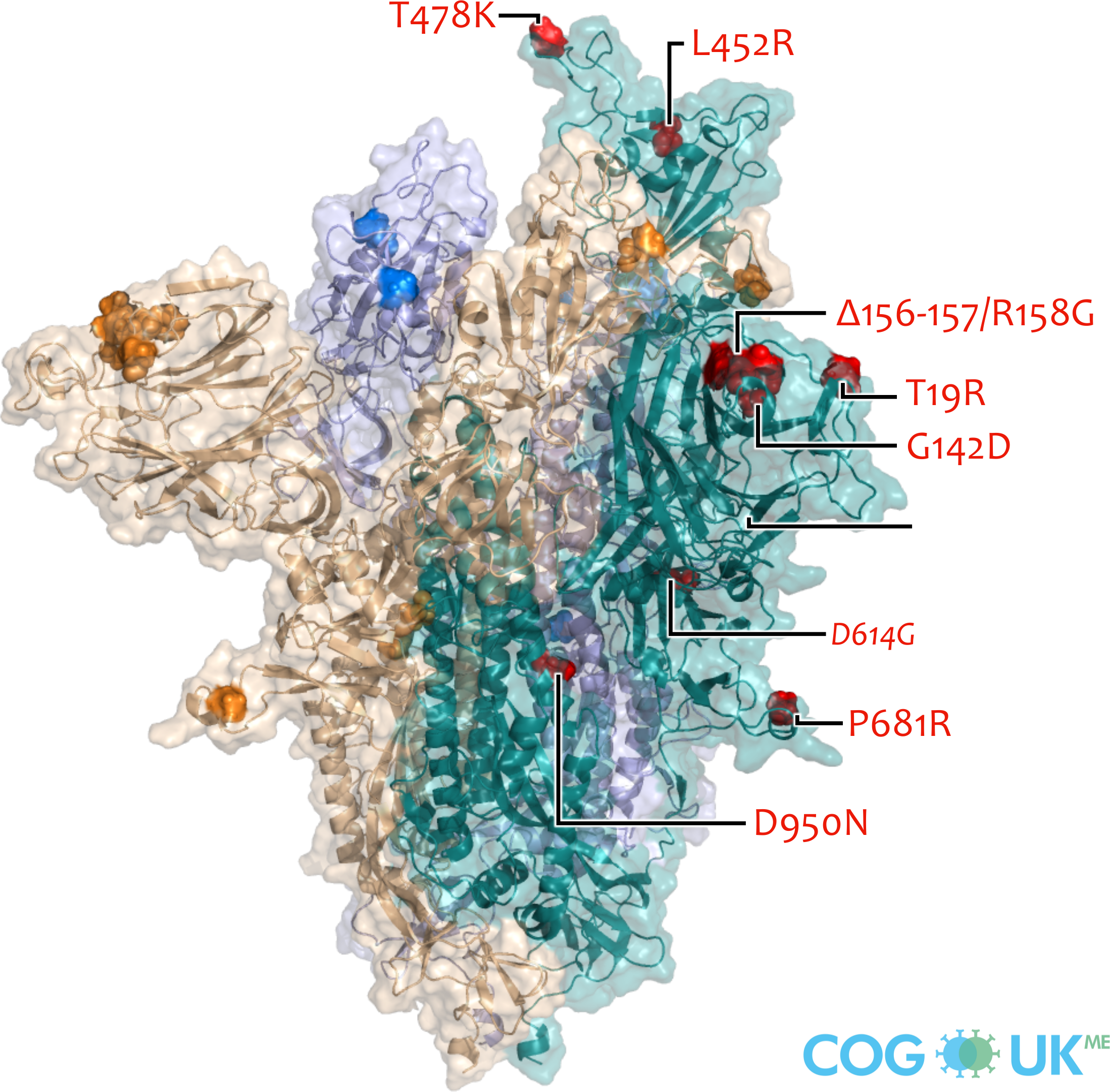
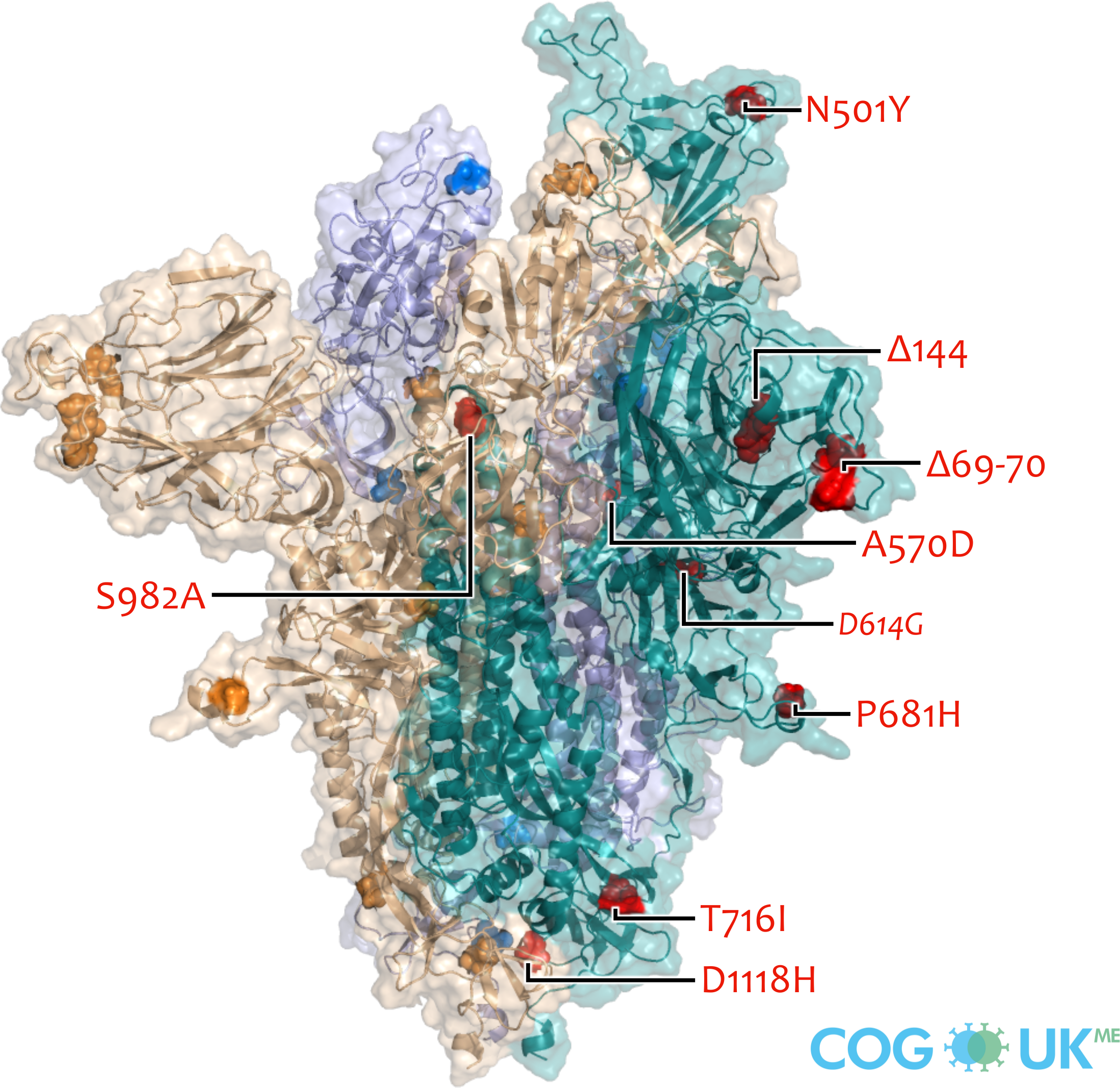
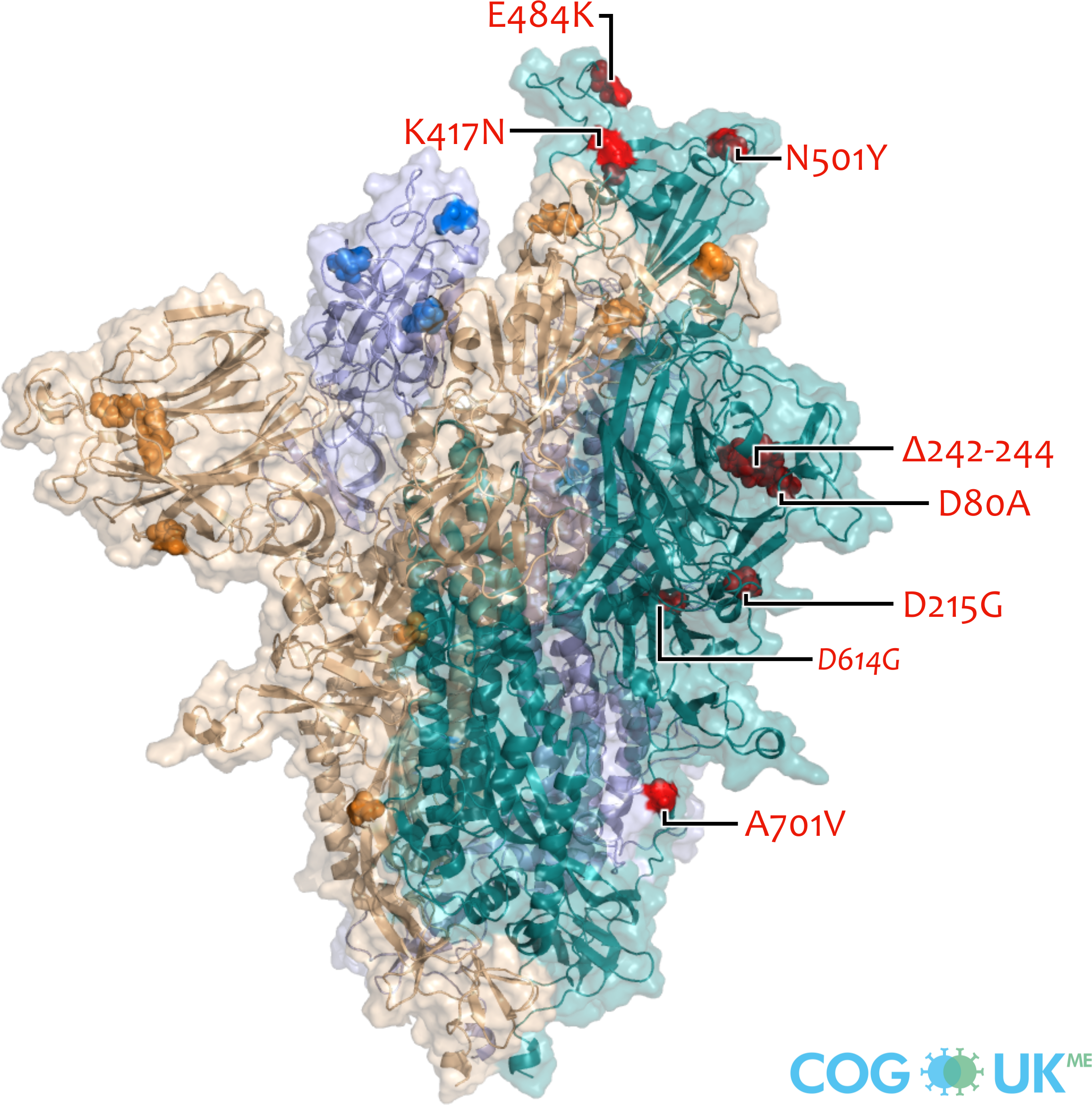
Spike protein structures showing locations of amino acid residues that are mutated in each variant of concern (VOC). The spike protein protrudes from the surface of the SARS-CoV-2 virus, is responsible for initating binding to and entry into host cells, and is also the primary target for antibodies that recognise the virus.
Each spike consists of three identical protein chains (shown in teal, blue and gold). Here, spike is shown in its 'open' conformation in which the receptor-binding domain of the teal chain is 'up' exposing the binding site that recognises the human ACE2 receptor.
On each chain, the locations of amino acid substitutions, deletions (Δ), and insertions (ins) which distinguish each VOC from the original genotype (Wuhan-Hu-1), are highlighted as opaque-surface spheres coloured in red (where they are labelled), blue and gold. The substitution D614G which is shared by common descent by all lineage B.1 descendants is italicised.
Visualisations are made using the ectodomain of a complete spike model (Woo et al., 2020) which is in turn based upon a partial cryo-EM structure (RCSB Protein Data Bank (PDB) ID: 6VSB (Wrapp et al., 2020)).
Spike protein mutations (Omicron: BA.1)
Spike protein mutations (Delta: B.1.617.2)
Spike protein mutations (Alpha: B.1.1.7)
Spike protein mutations (Gamma: P.1)
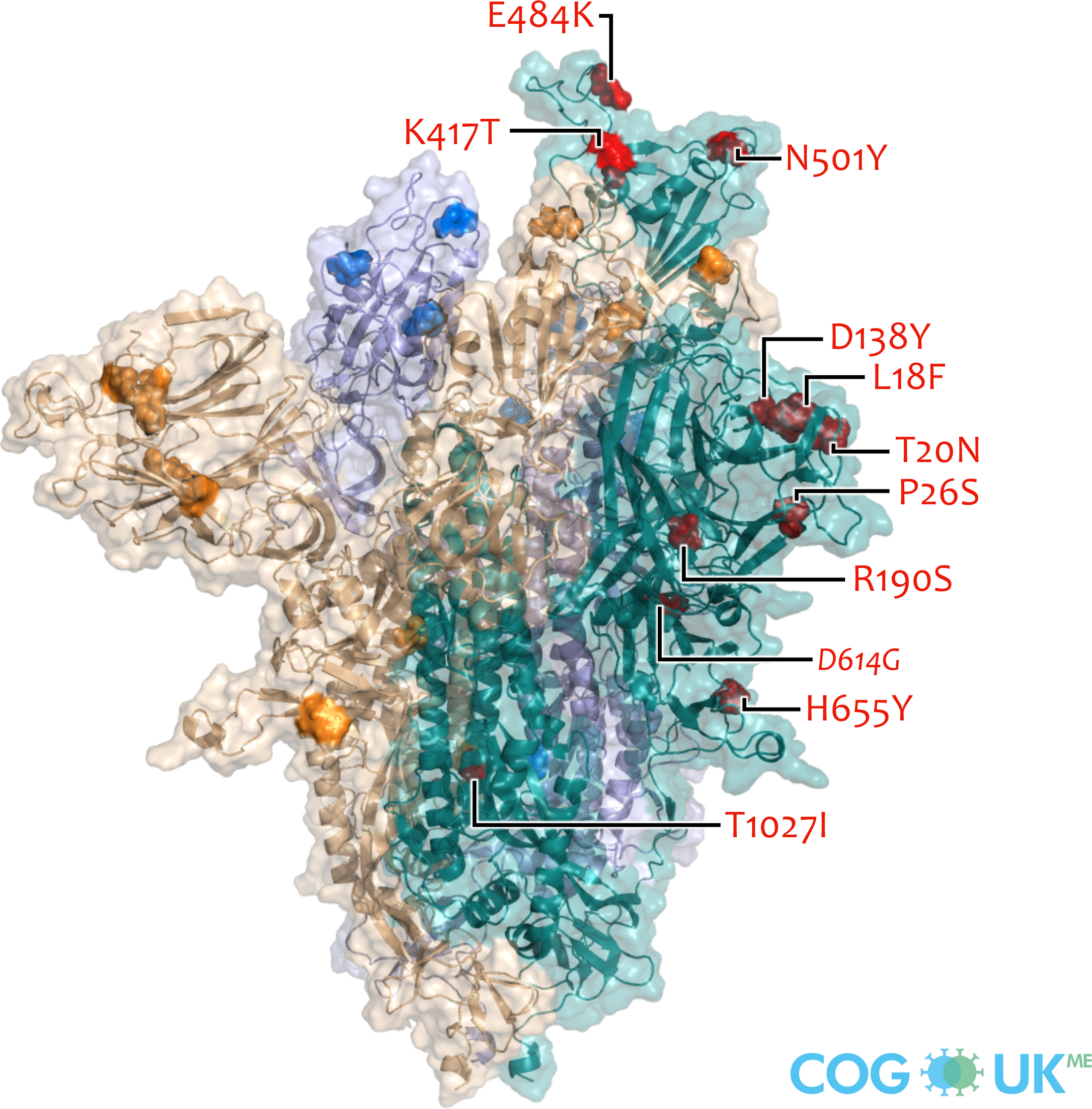
Spike protein mutations (Beta: B.1.351)
ESM-2 model scores and evolutionary selection signals of spike amino acid replacements
Structural context of spike amino acid replacements
Radar chart of spike amino acid replacements
Reference and query sequence of spike protein
List of spike mutations with relevant ESM-2 scores, selection analysis signals and amino acid properties
Prediction of amino acid replacements with high immune escape potential and evolutionary selection signals in the SARS-CoV-2 spike
ESM-2 (paper, repository) is a machine learning (ML) technique for natural language processing that has been found useful in assessing the effects of mutations on viral function and predicting mutations that may lead to viral escape. The model operates in an unsupervised fashion, meaning that it is trained to predict amino acids from the surrounding sequence context, with two components: grammar (or syntax) and meaning (or semantics). Semantic change corresponds to antigenic change, relative grammaticality captures viral fitness, and both high semantic change and relative grammaticality help predict escape potential.
The plot above displays the ESM-2 model scores of antigenic amino acid mutations present in the SARS-CoV-2 spike protein either in variants of concern (VOC) and variants under investigation (VUI) or for every possible amino acid mutation present in a given spike protein sequence (complete spike sequence) which is checked for mutations against the reference Wuhan-Hu-1 sequence. Mutations present in the spikes with available ESM-2 embedding scores are shown on the plot and listed on the above table. Filters are present to subset these mutations as needed or results can be refined further by filtering for mutations that were reported to confer antigenic change relevant specifically to antibodies or for amino acid replacements in T cell epitopes, based on published studies (see also 'Antigenic Mutations' tab).
In addition to the ESM-2 scores, signals of ancestral evolutionary selection in the animal (bat and pangolin) sarbecoviruses most closely related to SARS-CoV-2 (nCoV clade, defined in Lytras et al. 2022) are also displayed and can be overlayed on the spike protein structure (RCSB Protein Data Bank (PDB) ID: 6VXX (Walls et al., 2020)). Site-specific selection was inferred on a set of 167 sarbecovirus genomes, accounting for recombination by inferring selection separately in each non-recombinant segment (Martin et al., 2022). Briefly, sites under negative selection were inferred using the FEL (Kosakovsky Pond and Frost 2005) and sites under positive selection using MEME (Murrell et al. 2015) by testing on internal branches of the nCoV clade. Sites denoted as conserved have the same amino-acid residue among all sarbecovirus sequences in the analysis.
Evolutionary "diversity" of the site in the SARS-CoV-2 sequence was obtained as the entropy of the predicted distribution of credible evolutionary states. Structure-based epitope score (referred to as accessibility), which approximates antibody accessibility for each spike protein amino acid position, has been calculated using BEpro software (Sweredoski M., Baldi P., 2008).
About COG-UK/Mutation Explorer
Overview
The COG-UK/Mutation Explorer (COG-UK/ME) provides information and structural context on mutations and associated variants in the genes encoding SARS-COV-2 proteins that have been identified from sequence data generated by the COVID-19 Genomics (COG-UK) Consortium. We focus on SARS-CoV-2 spike gene mutations of potential or known importance based on epidemiological, clinical and/or experimental observations.
The Mutation Explorer comprises of:
- the designated global variants of concern and their structural contexts
- high frequency individual amino acid replacements, a subset of which may be important
- heatmap of antigenic mutations accumumulating on top of lineage-defining mutations of VOC/VUI
- frequency plots for mutations at specific residue in SARS-CoV-2 ORFs (Mutation Visualiser)
- mutations of potential antigenic significance as indicated by experimental studies: shown to lead to weaker neutralisation of the virus by convalescent plasma from people who have been infected with SARS-CoV-2 and/or demonstrated escape from some monoclonal antibodies (mAbs) that may be given to patients with COVID-19 (Antigenic Information: Antibody Sites)
- mutations in T cell epitopes as indicated by experimental studies (Antigenic Information: T Cell Epitopes).
Data source and processing
The analysis described in this report is based on 3,079,046 UK-derived genomes after dedeuplication, sequenced by COG-UK: complete data in the MRC-CLIMB database to 24/12/2024, with the latest sequence from 16/12/2024.
A report of the geographic distribution and prevalence of SARS-CoV-2 lineages in general, and global variants of interest, can be found here. Amino acid replacement, insertion and deletion counts for all SARS-CoV-2 genes in the global GISAID database can be found here.
Limitations
- This report is for information only. The clinical and public health importance of any single mutation, or combination of mutations cannot be determined from sequence data alone.
- Putative evidence for the importance of any single mutation, or combination of mutations can be derived from computational biology and further evaluated by laboratory experiments. Genomic and laboratory evidence then need to be combined with clinical datasets that are designed to allow detection of increased transmissibility, change in disease severity, drug resistance or altered vaccine efficacy. For this reason, surveillance and risk assessment of mutations and variants is a multi-agency process involving UK Public Health Agencies who have access to detailed information on patients and populations, and other groups including NERVTAG (New and Emerging Respiratory Virus Threats Advisory Group).
- COG-UK generates around 10,000 genomes a week, which will rise to 20,000 per week by March 2021. When COVID-19 infection rates are high, not all viruses from infected people will be sequenced and some mutations at low frequency will not be detected, but COG-UK aims to take representative samples from across the UK.
Background
Mutations arise naturally in the SARS-CoV-2 genome as the virus replicates and circulates in the human population. As a result of this on-going process, many thousands of mutations have already arisen in the SARS-CoV-2 genome since the virus emerged in late 2019. As mutations continue to arise, novel combinations of mutations are increasingly observed. The vast majority of mutations have no apparent effect on the virus. Only a very small minority are likely to be important and change the virus in any appreciable way. This could include a change in the ability to infect/transmit between people; a change in disease severity; or a change in the way the virus interacts with the immune system (including the response generated by a vaccine). We pay most attention to mutations in the gene that encodes the Spike protein, which is associated with viral entry into cells and it is relevant in the context of immunity and vaccine efficacy.
Glossary
- Mutation is used to describe a change of a nucleotide in the virus RNA genome, a subset of which results in a change in amino acid (sometimes referred to as a substitution or replacement), or a mutation can refer to a deletion or insertion event in the virus genome. By convention an amino acid change is written N501Y to denote the wildtype (N, asparagine) and replacement amino acid (Y, tyrosine) at site 501 in the amino acid sequence.
- Viral variant refers to a genetically distinct virus with different mutations to other viruses. Variant can also refer to the founding virus of a cluster/lineage and used to refer collectively to the resulting variants that form the lineage.
- Lineages are assigned combining genetic and, in the case of SARS-CoV-2 due to weak phylogenetic signals, also with epidemiological data. COG-UK uses the nomenclature system introduced by Rambaut et al. (2020), see https://cov-lineages.org.
- VUI is used by Public Health England to indicate Variant Under Investigation.
- VOC is used by Public Health England to indicate Variant of Concern.
Disclaimer
Dashboard reports are not advice. They capture research findings which are always necessarily provisional. They are for research use only. Commercial use/resale is not permitted.
How to cite COG-UK-ME
Please cite:
Derek W Wright, William T Harvey, Joseph Hughes, MacGregor Cox, Thomas P Peacock, Rachel Colquhoun, Ben Jackson, Richard Orton, Morten Nielsen, Nienyun Sharon Hsu, The COVID-19 Genomics UK (COG-UK) consortium, Ewan M Harrison, Thushan I de Silva, Andrew Rambaut, Sharon J Peacock, David L Robertson, Alessandro M Carabelli, Tracking SARS-CoV-2 mutations and variants through the COG-UK-Mutation Explorer, Virus Evolution, Volume 8, Issue 1, 2022, veac023, https://doi.org/10.1093/ve/veac023
Credits
COG-UK-ME has been developed as part of the COVID-19 Genomics UK (COG-UK) Consortium by Derek W Wright, William T Harvey, Joseph Hughes, MacGregor Cox, Thomas P Peacock, Rachel Colquhoun, Ben Jackson, Richard Orton, Morten Nielsen, Nienyun Sharon Hsu, Ewan M Harrison, Thushan I de Silva, Andrew Rambaut, Sharon J Peacock, David L Robertson and Alessandro M Carabelli (Wright et al. 2022) and uses data from the CLIMB-COVID framework (Nicholls et al. 2021). The dashboard is maintained by the MRC-University of Glasgow Centre for Virus Research. COG-UK is supported by funding from the Medical Research Council (MRC) part of UK Research & Innovation (UKRI), the National Institute of Health Research (NIHR) (MC_PC_19027), and Genome Research Limited, operating as the Wellcome Sanger Institute. We also acknowledge funding from the G2P-UK National Virology Consortium (MR/W005611/1), MRC (MC_UU_12014/12 and MR/R024758/1) and Wellcome Trust (220977/Z/20/Z). Follow COG-UK-ME and COG-UK to be notified of updates.
Contact Us
Get in touch with us at cvr-webresource-support@lists.cent.gla.ac.uk. To report issues with COG-UK-ME, use our issue tracker on GitHub.
Variants of concern (VOC) and under investigation (VUI) and any other variant by weeks and days
Variant sequence counts in the chart above are grouped either by week, starting on Sunday, or by day, and include counts of subvariants, except in the case of BA.5, where counts of the renamed subvariant BQ.1 have been subtracted. The most recent sequence data (approx. the last two weeks) have low sample numbers, so are highlighted with a grey box for the last two weeks of the weekly chart or from the second-to-last Sunday onwards for the daily chart.
Variants of concern (VOC) and under investigation (VUI) detected in the UK data
Variant sequence counts in the table below include counts of subvariants, where the variant names are denoted with .x, otherwise counts are for the specific variant names.
DISCLAIMER: COG-UK uses curated sequences for determining the counts of a given lineage. Other sources of information may be reporting cases with partial sequence information or other forms of PCR testing.
Download metadata
Download a CSV file, for each variant, containing COG-UK sequence name, sample date, epidemiological week, epidemiological week start date and global lineage. Cumulative UK sequences are filtered by the selected lineage of concern.
Download table
Download a CSV file comprising complete table data.
DownloadAntigenic amino acid replacements in variants of concern (VOC) and variants under investigation (VUI) in addition to their defining mutations
Amino acid replacement counts and percentages by week in the UK data
Mutation counts are grouped by week, starting on Sunday. The most recent sequence data (approx. the last two weeks) have low sample numbers so are highlighted with a grey box. When selecting Replacements, deletions are combined. When selecting Deletions, replacements are combined. Additionally, replacements or deletions with fewer than 5 sequences over all time in the UK are combined.
Amino acid replacements detected in the UK data: counts, percentages, nations and date of first detection
Individual amino acid replacements detected in UK genomes are shown (sequences ≥ 5). Neither insertions nor deletions, nor synonymous mutations are included.
NB Number of genomes is not equal to number of COVID-19 cases as data have not been deduplicated.
Deletions of nucleotides of lengths that are multiples of 3 in coding regions, in which substitutions are also tracked, are shown. Out-of-frame deletions that also result in a change in amino acid flanking the deletion are also annotated (e.g. ‘del156-157/R158G). Notation is relative to Wuhan-Hu-1.
Insertions of nucleotides of lengths that are multiples of 3 in coding regions, in which substitutions are also tracked, are shown. Insertions that fall between codons are labelled with the notation (e.g. 'Ins: 140-ITAL-141'). Insertions within codons are labelled (e.g. 'Ins: V120VL') which is a 3 nucleotide insertion within codon 120 which results in two residues ‘VL’ in the translated sequence. Notation is relative to Wuhan-Hu-1. Insertions containing ambiguous bases are not included.
Download metadata
Download a CSV file, for each amino acid replacement/del/ins, comprising COG-UK sequence name, sample date, epidemiological week, epidemiological week start date and global lineage. UK sequences are filtered by a 28 day period up to and including the most recent UK sequence date.
Download table
Download a CSV file comprising complete table data.
Spike amino acid replacements reported to confer antigenic change relevant to antibodies, detected in the UK data
The table lists those mutations in the spike gene identified in the UK dataset that have been associated with weaker neutralisation of the virus by convalescent plasma from people who have been infected with SARS-CoV-2, and/or monoclonal antibodies (mAbs) that recognise the SARS-CoV-2 spike protein (referred to below as "escape").
There is no evidence at the time of writing for this impacting on the efficacy of current vaccines or the immune response to natural SARS-CoV-2 infection.
Table key
Confidence
- High: Antigenic role of mutation is supported by multiple studies including at least one that reports an effect observed with (post-infection serum) convalescent plasma.
- Medium: Antigenic role of mutation is supported by multiple studies.
- Lower: Mutation is supported by a single study.
Spike protein domain definitions
- SP, signal protein (residues 1-13)
- NTD, N-terminal domain (14-303)
- RBD, receptor-binding domain (331-527) which includes the RBM, receptor-binding motif (437-508)
- FP, fusion peptide (815-834)
- Residues outside of these specific domains are labelled by subunit, S1 (residues 1-685) or S2 (residues 686-1173)
Download data
Download a CSV file containing COG-UK sequence name, sample date, epidemiological week, epidemiological week start date and global lineage. Cumulative UK sequences are filtered by the selected amino acid replacement.
Spike amino acid replacements in T cell epitopes, detected in UK data
T-cell epitope data have been compiled by Dhruv Shah, Sharon Hsu and Thushan de Silva, University of Sheffield.
Data are filtered depending on the experiments that have been used either showing 'Reduced T cell recognition' or through 'Epitope studies' (options on the left hand side).
Predicted binding percentile rank values have been calculated by Morten Nielsen, The Technical University of Denmark.
Table Key
- WT Percentile Rank Value and Mut Percentile Rank Value: predicted IC50 nM for the corresponding reported restricting allele. Predictions were performed using the NetMHCpan BA 4.1 algorithm, hosted by the IEDB.
- Fold difference indicates Increase/decrease in affinity defined by a two-fold difference in predicted IC50 nM.
- Binding is reported as a percentile rank value (as described here), the lower the value the stronger the binding.
- For HLA-I, values less then 2 are binders and values less than 0.5 strong binders.
- For HLA-II, values less then 5 are binders and values less than 1 strong binders.
T cell epitope sequence viewer
Move the slider to see sequence logs showing amino acid replacements in any epitope that overlaps on a specific position in the spike protein sequence. Each letter represents an amino acid replacement present in a specific epitope. The number below the sequence logo shows the position relative to the start position of the epitope. The height of a letter gives a measure of frequency of a mutation, whereas colour indicates amino acid chemistry. Frequencies are normalised within each epitope on a scale of entropy (0 to 4.3 bits). The wild-type epitope sequence and the start and end positions of the epitope are displayed above each sequence logo.
We thank Wagih, Omar. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics (2017) .
Download table
Download a CSV file comprising complete table data.
Genome browser showing T cell epitopes containing spike amino acid replacements, detected in UK data
The SARS-CoV-2 reference genome is displayed at the top, with the genomic sequence in the 4th row and the amino acid translations in the 2nd row. The other rows show a theoretical complementary strand sequence and alternative frame amino acid translations. Annotations for all genes are displayed in the annotations track. The epitopes track displays epitopes in the spike protein, in which amino acid replacements have been detected in COG-UK data. Click on an epitope to reveal details of amino acid replacements, including counts of sequences.
Spike profile expansion and contraction
Each spike profile is a set of amino acid substitutions listed relative to the original genotype (Wuhan-Hu-1). Spike profiles sampled within 7 days of the latest UK sequence are plotted below, with count of sequences in the latest 28-day period on the x-axis and a statistic estimating recent significant change in profile frequency on the y-axis.
Profiles of the Delta or Omicron variants of concern (VOCs) are described as amino acid substitutions relative to the core VOC profiles listed below.
Hovering the cursor over a point reveals the substitutions defining a spike profile, count in the latest 28-day period and associated pango lineages. The plot and table below can be re-drawn including data for each of the UK nations selecting a dataset below.
VOC core spike profiles: '+' indicates additional substitutions and '-' marks the absence of a substitution present in the core profiles below
Delta: T19R, G142D, Δ156-157/R158G, L452R, T478K, D614G, P681R, D950N
Omicron (BA.1): A67V, Δ69-70, T95I, G142D/Δ143-145, Δ211/L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F
Omicron (BA.2): T19I, L24S/Δ25-27, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K
Substitutions in VOC core profiles that are sometimes not identified VOC sequences (due to amplicon dropout during sequencing) are not listed as absent. For Delta profiles, G142D, is not listed as absent. For Omicron, due to widespread undercalling of core substitutions, the absence of core substitutions is currently not shown
Expansion/contraction: For each profile, i , the absolute value for this statistic is calculated using the observed frequency O i of profile i in each of the most recent 2-week periods j according to:
Spike profiles detected in the UK during the last week
Profile lists the amino acid substitutions (deletions and insertions are not currently included) in the spike protein relative to the original genotype (Wuhan-Hu-1). Note: Incomplete spike profiles may be called where the underlying sequence data is incomplete.
Amino acid substitutions is the count of spike amino acid substitutions relative to the orginal genotype (Wuhan-Hu-1).
Average growth rate (%) Calculated over the latest 56 day period to the date of the most recent UK sequence. The percentage change in frequency between each 2-week period within this period averaged and shown as a percentage.
Amino acid mutations reported to confer resistance to antiviral therapies, detected in the UK data
The table lists those mutations in the SARS-CoV-2 genome identified in the UK dataset that have been associated with resistance of the virus to antiviral treatments. There is variation in the detail of the viral assays between the different studies displayed here.
Combinations of spike amino acid substitutions that may confer resistance to antibodies in the therapeutical antibody cocktail, Ronapreve.
Plot showing the frequency of mutations affecting Ronapreve constituent monoclonal antibodies (mAbs) and their combinations (shown as lines) in cumulative UK SARS-CoV-2 genome sequence data. Spike amino acid substitutions known to affect either casirivimab or imdevimab mAbs were considered. The upper histogram shows the number of sequences per combination whereas the bottom left histogram shows the number of sequences with each specific substitution. Rows are coloured according to the mAb to which the greatest fold-decrease in binding was recorded (blue = casirivimab, orange = imdevimab), with a lighter shade indicating a fold-decrease of less than 100 and darker shade indicating 100 or greater.
The plot is generated using data from here.
Geographical distribution
Map showing the geographical distribution of variants, as either number of sequences or percentage relative to a specific region. N.B. Sequences without geographical information have been excluded from this analysis, so overall counts may be slightly lower than reported in VOCs/VUIs in the UK.
Possible effect of Omicron against mAbs
The table shows the fold reduction in neutralisation by monoclonal antibodies for circulating variants and single mutation spike profiles. Comparison between single mutation and full variant data indicates the role of each mutation in the evasion of the full variant. Some suggestion is made of the likely evasion profile of the Omicron (B.1.1.529) variant based on the mutations it contains. However, more definitive conclusions await clinical data and neutralisation assays involving the full virus.
Recombinant variants detected in the UK data
Recombinant lineage names do not contain information about their putative parental lineages and are named by their order of discovery: XA, XB, XC…, XAA, XAB,…XBA, etc.. Putative parental lineage information (which might be uncertain or incomplete) can be provided in the Pango lineage summary table. These numbers are an underestimate of the circulating recombinants due to detection issues.